


The rate at which a drug enters the body after administration is called the absorption rate, and is represented by the symbol ka. This is probably one of the simplest pharmacokinetic (PK) parameters to explain and understand. Let’s consider the case of oral administration first, then we can discuss other routes of administration. A drug is contained within a pill and is ingested by swallowing with liquid (usually water). The pill then moves through the intestinal tract (stomach, small intestine, large intestine) disintegrating along the way. The drug is then absorbed across the intestinal wall and into the portal vein. Eventually drug makes its way through the liver and into the systemic circulation. The absorption rate measures the rate at which drug moves from the intestinal tract into the systemic circulation.
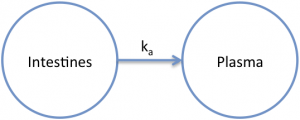
The absorption rate constant has units of inverse time. Most commonly, a first-order absorption process is used to describe the observed data. This means that the amount of drug that moves from the intestines to the plasma (systemic circulation) depends on the concentration of drug in the intestines. It is assumed that the drug concentration in the intestines is much higher than the concentration in the plasma. This assumption is often valid because of the large size of the systemic circulation (15 L) and the relatively small volume in the intestines (1-2 L).
Thus the absorption rate describes how the drug enters the body following administration. I have described a first-order rate constant here, but there are other models used to describe the absorption process such as zero-order (as is the case with extended-release tablets), Weibull (constantly changing absorption process), and bolus absorption. Each of these may have a different set of parameters to describe the absorption process, but in essence, they are replacing ka with a complex function. Different routes of administration generally follow different absorption profiles. For example, intramuscular follows first-order absorption, subcutaneous often follows zero-order absorption, and intravenous infusions generally follow zero-order absorption.
Drug developers frequently use in vitro-in vivo correlations (IVIVCs) to serve as a surrogate for human bioequivalence (BE) studies, support and/or validate the use of dissolution methods and specifications, assist in quality control during manufacturing and selecting appropriate formulations. Watch this webinar to learn about 2 important approaches to developing IVIVCs.
