


希少疾患の治療薬に関する規制状況は大きく変化しています。最近のいくつかの規制当局の諮問委員会の審議から、希少疾患の新薬開発の複雑性をあらためて垣間見ることができました。保健当局、医薬品開発者、キーオピニオンリーダーは、希少疾病を適応症とする臨床試験において意味のあるデータシグナルの帰属に苦慮しています。このブログでは、希少疾患開発の成功確率を最大化するために私たちが考慮すべき3つのポイントについてお話しします。デュシェンヌの例を用いて、いくつか特徴をご紹介していきたいと思います。
デュシェンヌ型筋ジストロフィー(DMD)は、進行性で致死的なX連鎖性の希少神経筋単発性疾患であり、治療選択肢はほとんどありません。DMD遺伝子の変異により、機能的なジストロフィンタンパク質の産生が阻害されることで発症します。変異のために、DMD患者は筋肉内で機能的ジストロフィンをほとんど産生しません。この機能的ジストロフィンの欠如がDMDの主な原因です。ジストロフィン経路を標的としている希少疾患研究者により、ジストロフィンに関連した薬力学に関する重要なデータがすでにいくつか公開されています。しかし、問題はジストロフィンのような標的関与バイオマーカーのレベルを上げることと、意味のある臨床的有効性との関連です。現在までに「ジストロフィン」ベースのアンチセンスオリゴヌクレオチド(EXONDY 51、VYONDY 53、AMONDY 45、VILTEPSO™)と「マイクロジストロフィン」AAV療法(ELEVIDYS)が早期承認されています。
Ensure that your biomarker is biologically plausible.
バイオマーカーやサロゲートエンドポイントの選択は、以下の2つの理由から希少疾患プログラムを成功させるために非常に重要です。(バイオマーカー戦略の詳細については、このホワイトペーパーをお読みください)。 1つ目は、選択したバイオマーカーと臨床エンドポイントとの間に予測可能な関係を確保すること。2つ目は、薬物反応の変化が生物学的または生物学的妥当性によって説明されることです。もしあなたがよく知られた代用エンドポイントがあるプログラムに取り組んでいるのであればラッキーです。しかし代用エンドポイントがない場合は、生物学的妥当性を証明しなければなりません。DMDが神経筋疾患であることから、遺伝子治療薬の開発者は、FDAの神経変性疾患のためのヒト遺伝子治療ガイダンス(FDA 2022)に記載されている構成を利用することができます:「例えば、重篤な神経変性疾患の原因となる、よく理解され、十分に文書化された単発性変化を直接標的とする【遺伝子治療】GT製剤のような特定の状況下では、代替エンドポイントの使用が適切な場合があります。」
DMDのほとんどすべての治療法には、ジストロフィンがバイオマーカーとして含まれています。骨格筋と心筋の両方で欠損したジストロフィン遺伝子を置換することで、DMD患者のQOLが向上すると言われているからです(Davies et al., 2019)。遺伝子治療開発において、組換えアデノウイルス関連ウイルスの低い遺伝子容量とジストロフィンmRNAの大きなサイズのマッチングは困難でした。今回のELEVIDYSの承認により、マイクロジストロフィンも治療上重要であることが示され、DMDに対する遺伝子治療の選択肢が増える可能性があります。バイオマーカーが臨床エンドポイントとどのように相関するかは極めて重要であり、このような新規バイオマーカーを含む研究は、バイオマーカーと臨床エンドポイントとの関係を明確にする必要があります。例えば、DMDに関するFDAのガイダンスでは、NSAA(North Star Ambulatory Assessment)を使用して粗大運動機能を測定することを提案しています。その他の臨床的測定としては、歩行可能な3歳以上の小児を対象とした、階段4段昇降時間や10メートル歩行・走行時間などの計時機能検査などがあります。
ランダム化比較臨床試験として最適な臨床プログラムを実施
新薬開発プログラムにおいて、新規治療へのシグナル帰属は常に重要です。薬剤が意図したとおりに作用しているか?これは生物学の問題であるだけでなく、その生物学がどのように評価されたかという問題でもあります。スポンサーは常により簡単で安価な臨床試験の方法を探り、対照薬のないオープンラベル試験を選択することがあるでしょう。このような意思決定は新薬候補の肥満臨床試験をプラセボ群なしで行うようなものです。想像してみてください、もしその試験にプラセボがあったとしたら、プラセボ群では試験参加者全員に食事療法と運動療法が要求されるため、体重が減少していることがわかるでしょう。もし臨床試験デザインが適切でなかった場合、そのシグナルを治療によるものと結論づけるかもしれません。したがって、一般的な疾患であれ希少疾患であれ、新しい分子の有効性と安全性を評価するために無作為化臨床試験(RCT)を用いることは、常に安全な選択です。RCTはオープンラベル設計よりも統計学的に優れており、DMDに関するFDAの業界向けガイダンス (2018)でも強調されています。
臨床プログラム内で薬理学的なばらつきが大きい場合(例えば、年齢が2歳から25歳まで等)、活性に関する推論を行うために必要なサンプルサイズがないことに気づくかもしれません。無作為化試験だけでなく、オープンラベル試験も組み合わせることができます。一方、サブセット解析に頼ることもできます。
サブセット解析にはメリット・デメリットの両方があります。メリットとしては、仮説を立てることができる点です。しかし、仮説を確認するためにサブセット解析を使用する場合、統計的に有意な偽陽性の結果が出る確率が高くなるリスクがあります。調査するグループが多ければ多いほど、偶然に統計的に有意な効果が見つかる確率は高くなります。 (“p-hacking”; Head et al., 2015). サブグループ解析やプールデータに基づいて仮説検定を行うには、仮説検定と多重度調整を事前に指定します。米国FDAは、探索的サブグループ解析は有効性の確認には不十分であると明言しています。
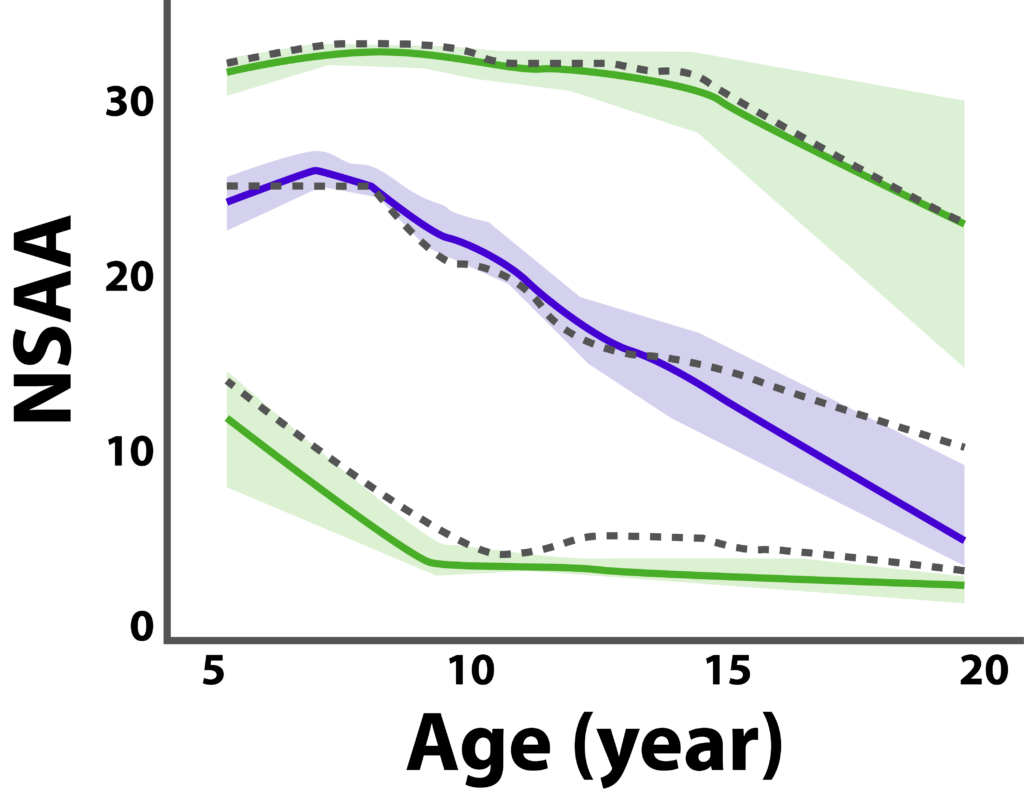
Lingineniらは、臨床試験デザインを最適化するために、モデルを活かしたアプローチを使って、DMDの注目すべき共変量と疾患進行情報を同定しました。図 2は、DMD試験における一般的なエンドポイントであるNSAAのプロフィールを示しています。視覚的予測チェックは、観測データの中央値(黒の破線)と第5パーセンタイルと95パーセンタイル(それぞれ下側の破線と上側の破線)を示し、青い網掛け部分は中央値のモデル予測値の 90% CIを示し、緑の網掛け部分は第5パーセンタイルと95パーセンタイルのモデル予測値の 90% CIを示します。実線はモデルによる予測を示しています。
臨床プログラムにて外部対象(External Control)を活用する
臨床開発プログラムにおける外部対象試験への規制当局の関心が高まり、受け入れられつつある一方で、その活用はしばしば多様であり、ベストプラクティスと矛盾しています。従って、このような対象の使用はしばしば裏付けと見なされます。外部対照を用いた統合解析を使用している希少疾患開発者は、試験レベルおよび統合解析においてこのようなアプローチを使用しています。傾向スコア加重法を用いて、臨床試験に登録された被験者と類似性の高い外部対照被験者を選択するものもあります。このような外部対照試験データは通常、自然史コホートのライブラリーから入手します。
DMDの場合、スポンサーは通常、Cooperative International Neuromuscular Research Group (CINRG)のDuchenne Natural History Study (DNHS)またはFinding the Optimum Regimen for Duchenne Muscular Dystrophy (FOR-DMD)の臨床試験から必要なデータを入手します。現在進行中のNH試験も多数あります(NCT03882827など)。ここでの課題は、適用される組み入れ基準と、スポンサーの薬効試験に登録された被験者の人口統計学的特徴との間に整合性を見出すことです。
外部対象の比較には限界もあります。たとえば、疾患経過の不均一性と非比較性の可能性、意図された治療効果の特徴づけにおけるバイアスの役割、外部集団と試験集団の真の類似性などです。ここでの焦点は、未観測または定量化不可能なベースライン特性です。傾向スコア加重法のようなものも、交絡因子に関する検証不可能な仮定に左右されます。
モデルを活かした医薬品開発(MIDD)が、希少疾患治療の針をいかに有意義に動かすことができるかについての詳細は、Journal of PK and PDに掲載された著者の論説をご参照ください。
参照文献
- Davies KE et al. Micro-dystrophin Genes Bring Hope of an Effective Therapy for Duchenne Muscular Dystrophy. Molecular Therapeutics. 2019 Mar 6; 27(3): 486~488.
- FDA Guidance, Duchenne Muscular Dystrophy and Related Dystrophinopathies: Developing Drugs for Treatment Guidance for Industry, February 2018.
- Head ML et al. The Extent and Consequences of P-Hacking in Science. PLoS Biology. 2015 Mar; 13(3): e1002106
- Lingineni K, Aggarwal V, Morales JF, et al. Development of a model-based clinical trial simulation platform to optimize the design of clinical trials for Duchenne muscular dystrophy. CPT Pharmacometrics Syst Pharmacol. 2022;11(3):318-332.
- Wang H et al. Clinical Trials with External Control: Beyond Propensity Score Matching. Statistics in Biosciences. 2022, 14, 304–317.

