


The era of gene therapy may have started a couple of decades ago, but approvals of agents based on the platform have been relatively recent. In 2017, Spark Therapeutics, Inc. received FDA approval of voretigene neparvovec-rzyl (Luxturna™), a recombinant adeno-associated virus serotype 2 (AAV2) vector expressing the gene for human retinal pigment epithelium 65 kDa 5 protein (hRPE65), for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. In 2019, AveXis, Inc. received FDA approval for licensure of onasemnogene abeparvovec-xioi (Zolgensma™), an AAV vector-based gene therapy indicated for the treatment of pediatric patients less than 2 years of age with spinal muscular atrophy (SMA) with biallelic mutations in the survival motor neuron 1 (SMN1) gene. These approvals have generated significant interest and enthusiasm for these next generation therapeutic strategies. Despite the promise of gene therapy, there are important limitations related to the episomal expression constructs on which the technology is based. It is against this backdrop that the advent of gene editing appears most intriguing, especially given the theoretical advantage in durability over episomal gene therapies in rapidly growing tissues.
Additionally, gene editing technologies have the potential to offer almost limitless opportunities to advance precision medicine for unmet medical needs. Indeed, the “Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)”-Cas platform has been a center-stage story making the headlines in novel ways to advance therapeutic discovery. What better way for genetics, cellular and molecular biology, and pharmacology as three strategic pillars of modern medicine to come together in the research and development of treatments for genetically validated rare diseases? So, what is CRISPR-Cas9 technology? If one surveys the recent publication literature, it is replete with references to this tool. In the most simplistic sense, CRISPR is a new technology that allows one to edit or delete a genomic region in intact cells and organisms. Of such prominence is this finding that two scientists, Emmanuelle Charpentier and Jennifer Doudna received the Nobel Prize in 2020 for the development of this method for gene editing.
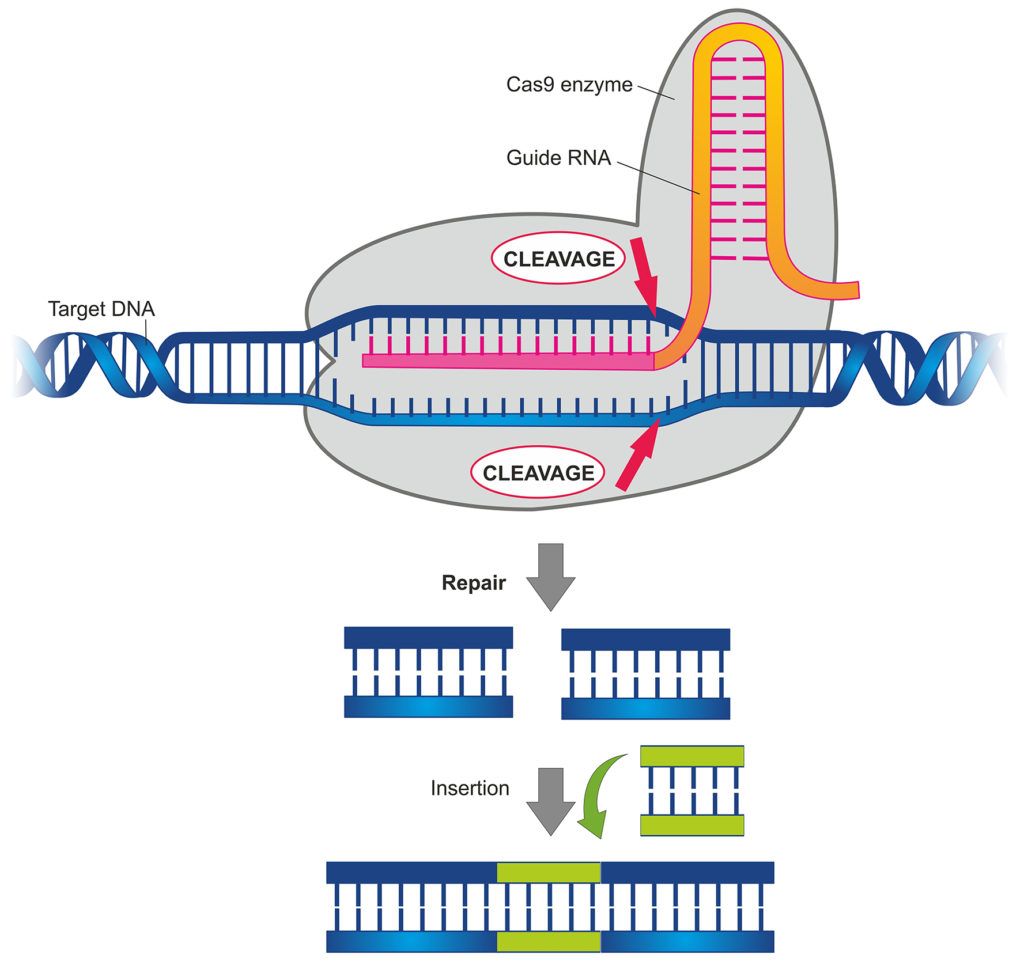
How CRISPR/Cas9 technology can be used for gene editing
Some of the therapeutic advances include using CRISPR-Cas9 in hematopoietic stem cells to address severe hematologic conditions like sickle cell disease and beta-thalassemia. In this case, CRISPR-Cas9 was introduced into bone marrow-derived cells in an ex vivo manner, and edited cells capable of producing functional hemoglobin were reintroduced into patients (1). A CRISPR-Cas9-based approach in genetically correcting the CEP290 mRNA was also shown to be effective in retinal dystrophy disorder (2). In this regard, the developed therapeutic, called EDIT-101 is being administered by a subretinal route leading to restoration of functional CEP290 RNA (3). In neurodegenerative diseases, for example, the potential for gene editing therapeutics is boundless if challenges related delivering the therapeutic components of the CRISPR-Cas9 system are addressed. Another example of a therapeutic approach using CRISPR-Cas9 is Intellia’s NTLA-2001, an in vivo gene editing agent consisting of a lipid nanoparticle that contains messenger RNA encoding Cas9 and a single guide RNA targeting the transthyretin (TTR) gene that is being investigated for treating ATTR amyloidosis. In a small Phase 1 patient series, NTLA-2001 was shown to reduce serum TTR levels thus providing optimism for in vivo CRISPR-Cas9–mediated gene editing as a potential therapeutic strategy (4).
Four main stories emerge from these examples of gene editing therapeutics. One, a novel and unique way is being implemented for delivering gene editing therapeutics. This has implications in the delivery of the therapeutics and the need for it to be “bioavailable”. Second, the safety of gene editing therapeutics is being scrutinized, particularly where off-target aspects are envisioned. Third, the ideal target population must be carefully considered, especially in an environment where episomal gene therapies may be available. Fourth, and perhaps the most important, is selecting the right dose. When small molecules, and to some extent, simpler biologics are considered, there is often a dose/response or an exposure/response relationship that signifies an underlying physiologically sensible assumption. This means that there is usually a broad pharmacodynamic dose range. However, in gene editing therapeutics that correct a certain course of action by permanently changing the genome of a particular tissue, there is an explicit need to ensure the right dose is selected the first time.
Let’s examine these four key areas in more detail.
Translational strategies
Delivery of vectors presents a proximal challenge in ensuring optimization of the availability of the therapeutics at the site of action. In a traditional sense. when we think about small molecules, we visualize the central systemic compartment and the deeper tissue site of action compartment, wherein there is a determined kinetic sequence leading to availability of the drug at the site of action. The concept is similar for gene editing therapeutics; however, the focus is now on the vector delivery.
Commonly cited options for delivery of gene editing components include viral vectors and lipid nanoparticles (LNPs). An adeno-associated viral vector comprises the capsid, which contains genetic material (for use in gene editing or gene therapy platforms) and the promoter (in the case of episomal gene therapies). In this context, capsid selection and optimization is key because it dictates the potency of the therapeutic, its immunogenicity, but also the degree of specificity (i.e., tropism) to the chosen tissue of interest. The literature contains much debate on the functionality of the chosen vector profiles, as it relates to intracellular transport, nuclear entry and uncoating, all of which can influence the level of functional transduction. Ensuring vectors are engineered in the context of delivery and optimization is a critical consideration. Patients will typically have to be screened for pre-existing immunity to the specific AAV vector being used for delivery. If there are pre-existing anti-AAV antibodies to the serotype being used, often transduction will not be successful. Given treatment-emergent immunity, dose selection is critical as re-dosing is not possible.
Lipid nanoparticles provide a differentiated option for delivering nucleotide-based material to support gene editing or gene therapy approaches. LNPs are clinically validated as an effective and safe delivery modality to the liver. Unlike AAVs, LNPs are very efficient in transfecting hepatocytes, and there is no de novo limiting immunity, so re-dosing is possible. LNPs are engineered to be metabolizable, limiting exposure to non-natural lipids and the gene editing cargo, which minimizes risk of genotoxicity and immunogenicity. Intensive discovery efforts are currently underway to enable LNPs to target other tissues.
Minimizing off-target activities
There has been considerable debate in the field on the off-target effects of gene editing therapeutics, as it relates to single gene editing. One risk relates to the possibility of a single guide RNA (gRNA) to edit unintended sites. This could lead to off-target mutagenesis, which may involve permanent alteration of genes and their function. Consequently, there is an emphasis in the field on the need for ensuring the highest level of selectivity and specificity for guide RNAs.
Target population
For episomal gene therapies, there is appropriate concern is around the durability of efficacy if the construct is administered early in life. The problem is that there will be loss of the episome over time if it is dependent on expression in growing and dividing cells (episomes will not be duplicated in dividing cells and are typically lost in the process of cellular proliferation). On the other hand, gene editing will result in a permanent change to the genome that will be passed along to the daughter cells upon cell division. Hence, gene editing may be more suitable for pediatric uses that require lifelong expression in dividing tissues.
Dose selection challenges
Unlike small molecule and simpler biologics development, the range of doses to be studied with gene editing platforms is relatively well circumscribed. The dose range is restricted by the safety and tolerability of the vectors in which the gene editing components are delivered, with liver injury typically limiting the amount of lipid nanoparticle and adeno-associated viral vector that can be administered. In that regard, it is essential to ensure sufficient translation exists in the extent of restored function with the therapeutic. This threshold is defined by the unmet medical need the therapeutic is supposed to address. Because the therapeutic range is book-ended by transgene production on one hand and the potential for toxicity on the other, the choice of doses to be tested in clinical trials need to be carefully decided. Furthermore, since administration of adeno-associated viral vectors results in robust generation of anti-capsid antibodies, there may be only “one shot on goal” for such a therapeutic.
To assist in dose selection for adeno-associated viral vectors, there needs to be an understanding of the vector shedding and biodistribution properties, which can be achieved by measuring the DNA in the tissues of primary interest as well as in whole blood. An understanding of biodistribution of viral vectors has emerged from the episomal gene therapy experience. Such an approach was used for the approval of voretigene, wherein this measurement was performed in tears from both eyes, and from serum, and whole blood of patients in the Phase 3 study. It is instructive to note that in the Luxturna summary basis of approval, the Phase 1 study included 12 patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy that received three doses, with no apparent dose related findings. The highest dose (1.5 x 1011 vector genomes) was then chosen for the Phase 3 study. Similarly, in the Zolgensma summary basis of approval, vector shedding was studied at multiple time points, and data showed that the vector DNA was shed in saliva, urine, and stool after infusion of study treatments. In their phase 1 program, two doses were studied with clear dose-response relationship with respect to efficacy described. Patient cohorts received 4.3×1013 to 4.6×1013 vg/kg and 1.1×1014 to 1.4×1014 vg/kg. The chosen Phase 3 dose was 1.1 × 1014 vg/kg dose.
There is considerable room for including model-informed drug development (MIDD) approaches for both gene therapy and gene editing, and a recent review by the FDA authors emphasizes these aspects (5). The authors note that MIDD would help with intracellular trafficking and transgene clearance, aspects that are poorly understood with these newer therapeutics and should be the continued focus of exploratory investigations.
Certara is a world leader in model-informed drug development. Please contact us for any complex biologics expert consulting needs.
David Gutstein, MD is a vice president at Regeneron with keen interest and specialization in gene-based therapeutics and is a guest contributor to this blog. Dr Gutstein’s research includes the Regeneron collaboration with Intellia in gene editing focusing on NTLA-2001 and other programs.
参照文献
- Frangoul H et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med. 384:252-260 (2021).
- den Hollander AI et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet.79:556–561 (2006).
- Maeder ML et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25, 229–233 (2019).
- Gillmore JD et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med. 2021 Jun 26. doi: 10.1056/NEJMoa2107454. Online ahead of print.
- Belov A et al. Opportunities and challenges for applying model-informed drug development approaches to gene therapies. CPT Pharmacometrics Syst. Pharmacol. 10:286–290 (2021).
