


生物学的同等性(BE)は、バイオファーマコエンティクスにおいてよく誤解される用語です。For starters, bioequivalence is a technical term baked in regulation. The Code of Federal Regulations (CFR) Subchapter D deals with drugs for human use, and 320.1 contains the key definitions regarding this subject. These are captured in full as follows:
§ 320.1 Definitions.
Bioavailability is defined in (a.) as:
Bioavailability means the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action.
Bioequivalence is defined in (e.) as:
Bioequivalence means the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study.
Bioequivalence means the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study.
That is one heavily nuanced definition. However, as with all things legal, interpretation is highly contextual. It doesn’t say to show bioequivalence within the confidence bounds of 0.8 and 1.25, and nowhere does it show that the dose of the test or reference should be identical. Before we launch into the myths, let’s try to understand the scientific basis behind the phrase “absence of a significant difference” which is so important in bioequivalence.
The United States FDA considers two products to be bioequivalent if the 90% confidence intervals of the geometric mean test/reference Cmax and AUC ratios fall within the bioequivalence confidence intervals of 80-125%. To obtain geometric means, the pharmacokinetic data are log-transformed prior to conducting an analysis of variance, then back-transformed before calculating the test/reference ratios.
Geometric mean ratios and log-transformed data are used to test the hypothesis that the 90% confidence interval of extent (total exposure, area under the plasma concentration time curve from pre-dose to the last measurable time point, AUC0 to last extrapolated to infinity) and the maximum concentration (peak exposure, the single point estimate of maximum observed concentration in the plasma concentration time curve) fall within the acceptance limits of 80–125%.
What constitutes a clinically acceptable change has been debated. Thresholds of clinical relevance in the order of a 20–25% change has been discussed widely (Williams et al., 1976). There was this 75/75 rule that stated that two formulations are equivalent if at least 75% of the individuals being tested had ratios (of the various pharmacokinetic parameters obtained from the individual results) between the 75% and 125% limits, and the study had the statistical power to detect a small 20% difference between the two formulations. In 1992, the FDA published the guidance on “Statistical Procedures for Bioequivalence Studies Using a Standard Two-treatment Crossover Design.”In this document, the recommended statistical approach was based on average bioequivalence and underpins the assessment of BE based on the confidence interval approach.
Let’s reflect now on some frequent bioequivalence myths.
Myth 1: I must bridge my formulations before using a new capsule in Phase 3. I need a BE study, and I need to use “BE bounds”.
This is a very common misunderstanding. The expectation for bioequivalence is when the formulation that is taken into a pivotal registrational study (e.g., Phase 3) undergoes further formulation changes during or after phase 3 and before commercialization. A bioequivalence study is necessary at this stage if the variance around the changes made to the formulation were to deviate as specified in the SUPAC IR guidance (FDA, 1995).
It is widely accepted that formulations will evolve during the IND stage of development, such as moving from a drug in bottle suspension from Phase 1 to a capsule in Phase 2. For these “internal” bridging studies, just a relative BA study is needed to determine whether to dose adjust before taking the new formulation into the next study. No BE study or meeting BE bounds are necessary.
Myth 2: I am an innovator company. I have a product with a wide therapeutic window. I know regulatory agencies require BE bounds to be met for my product.
If you are an innovator company, you will have valuable data on the PK, exposure-response, safety, and efficacy of your product. All these data can be leveraged to justify the clinical meaningfulness of any PK differences seen in your BE studies.
FDA guidance explicitly calls out approaches when BE studies fail: “When BE is not demonstrated, the sponsor should demonstrate that the differences in rate and extent of absorption do not significantly affect the safety and efficacy based on available dose-response or concentration-response data. In the absence of this evidence, failure to demonstrate BE may suggest that the test product should be reformulated, or the method of manufacture for the test product should be changed, or additional safety or efficacy data may be needed for the test product.”(FDA, 2014).
One common mitigation is to power the study for BE bounds and then justify the reasons for failure, if possible, versus being creative with bounds.
Myth 3: I am doing a food effect study with my new reformulated dosage form, and I need to use BE bounds as a criterion of clinical significance.
A food effect study shouldn’t be confused with a fed BE study. A fed BE study is required for generic product approval in addition to a fasted BE study. A food effect study is typically performed in the new drug application package for a new molecular entity. As such, it is a descriptive study and serves to understand the effect of a standard high fat breakfast on the performance of the dosage form simulating a “worst case scenario.”Thus, powering a study for BE criterion is not required.
Myth 4: I have failed f2 in multi-media dissolution test. I can use modeling to show the differences are not clinically meaningful. I will not need to demonstrate BE.
When comparing two formulations for bioequivalence, the first in vitro analytical test used to assess whether the two formulations are meaningfully different is the similarity factor, called “f2” (Krishna and Yu, 2007). F2 is simply a measure of the similarity of the dissolution profile between the test product (usually a newer or revised formulation) and the reference product (usually a product that has been used until now, or the innovator product when considering generic equivalent). Such a test is done under exploratory contexts or within acceptable regulatory contexts. To adhere to the latter bar, pay attention to the volume and type of dissolution media, rpm conditions, and defined sample size to meet the multi-media dissolution conditions. Then sample the media to measure drug amount dissolved to generate a dissolution curve for the products. Finally, use the data to calculate the f2.
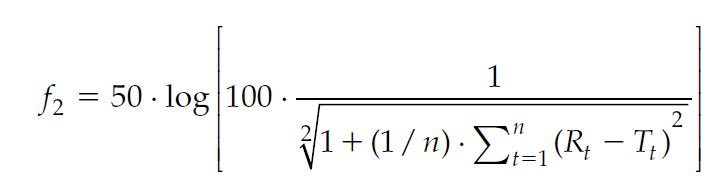
In the above referenced equation, f2 is the similarity factor, Rt and Ttare the cumulative percentage dissolved at each selected time point for the reference (R) product and test (T) product. Dissolution profiles are considered similar when the f2value is ≥50.
In general, f2 failure (any value of f2 that is <50) triggers an in vivo BE study. If f2 fails, the probability of meeting bioequivalence is usually low. At this stage, many sponsors will improve the dosage form performance. Sponsors can look at the formulation design space and re-adjust accordingly. The principles of model-informed drug development can still be used to rationalize that the changes might not be clinically meaningful.
Myth 5: I changed my formulation from that used in phase 3. I can market my new formulation without running a BE study.
When you change a formulation meaningfully (i.e., as specified in SUPAC guidances) from that used in Phase 3, you need to perform a sufficiently powered BE study to show that the formulations are not meaningfully different. If the BE study fails, then it becomes an approvability issue, if the differences are not readily justifiable as not being clinically meaningful. There is considerable room to renegotiate this scenario using integrative analyses and tools.
Be aware of certain post-approval changes that may trigger a BE study as well (e.g., SUPAC IR). If there are certain major changes in components, composition, manufacturing site, and/or method of manufacture after approval, the FDA recommends demonstrating in vivo BE for the drug product after the change in comparison to the drug product before the change.
Certara scientists have expertise in the use of model-informed drug development tools as well as the use of physiologically-based pharmacokinetic (PBPK) modeling as it pertains to biopharmaceutics.
To learn more about using PBPK to demonstrate bioequivalence, please read this white paper:
ホワイトペーパー
References and recommended reading
Food and Drug Administration (1992). Guidance for industry: statistical procedures for bioequivalence studies using a standard two treatment crossover design. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (1995). SUPAC-IR: Immediate-Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (1999). Center for Drug Evaluation and Research (CDER). Statistical Information from the June 1999 Draft Guidance and Statistical Information for In Vitro Bioequivalence Data, Posted on 1999年8月18日. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (2001). Guidance for industry: statistical approaches to establishing bioequivalence. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (2003). Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations. US Department of Health and Human Services, Rockville, MD.
Food and Drug Administration (2014). Guidance for industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs — General Considerations. US Department of Health and Human Services, Rockville, MD.
Krishna R, Yu L (2007). Biopharmaceutics applications in drug development, Springer, NY.
US Code of Federal Regulations. (2000)。Bioavailability and bioequivalence requirements. US Government Printing Office, Washington, DC, Vol. 21, Part 320. Also see: https://www.govinfo.gov/content/pkg/CFR-2009-title21-vol5/pdf/CFR-2009-title21-vol5-sec320-1.pdf. Williams RL et al (1976). Where are we now and where do we go next in terms of the scientific basis for regulation on bioavailability and bioequivalence? FDA Biopharmaceutics Coordinating Committee. European Journal of Drug Metabolism & Pharmacokinetics 25, 7 – 12.

