


抗体薬物複合体(ADC)は、腫瘍細胞を消滅させるユニークな方法であり、単独療法であれ併用療法であれ、腫瘍学において十分に利用されていない免疫治療薬の選択肢です。ADCの最初のFDA承認は2011年でした...
ADC development is inherently challenging because it requires close collaboration between the “biologic” and “small molecule” mind-sets. In many ways, the fusion of the two inherently varied disciplines is required to ensure the best thinking supports an ADC development program. Probably the best modern-day rendition of Paul Ehrlich’s magic bullet concept, administration of the ADC results in the antibody binding to the antigens on the surface of tumor cells. The complex with the antigen is soon internalized within the cell. The cytotoxic payload is then released from the antibody causing cell death. Depending on the type of linkers used, there may be an additional step of lysosomal proteolysis before apoptotic cell death. ADC engineering represents advanced molecular science that needs careful consideration of upstream development, some of which I will reflect upon in this blog.
Enabling smart discovery
The first consideration in ensuring the identification of a viable ADC is in the discovery phase. In traditional development, one encounters a skill-focused, linear sequencing of expertise, from discovery to development. However, the discovery of an ADC is perhaps more unique in that some of the downstream consequences of the asset must also be carefully considered. It is critical to ensure the right components are assembled, as it relates to the monoclonal antibody, the linker, and the payload. Once the asset is identified, it becomes quite tricky to re-do this assembly. The choice of the linker is as important as the payload. Often, more focus is given to the payload than to the linker itself and this could result in considerable development delays.
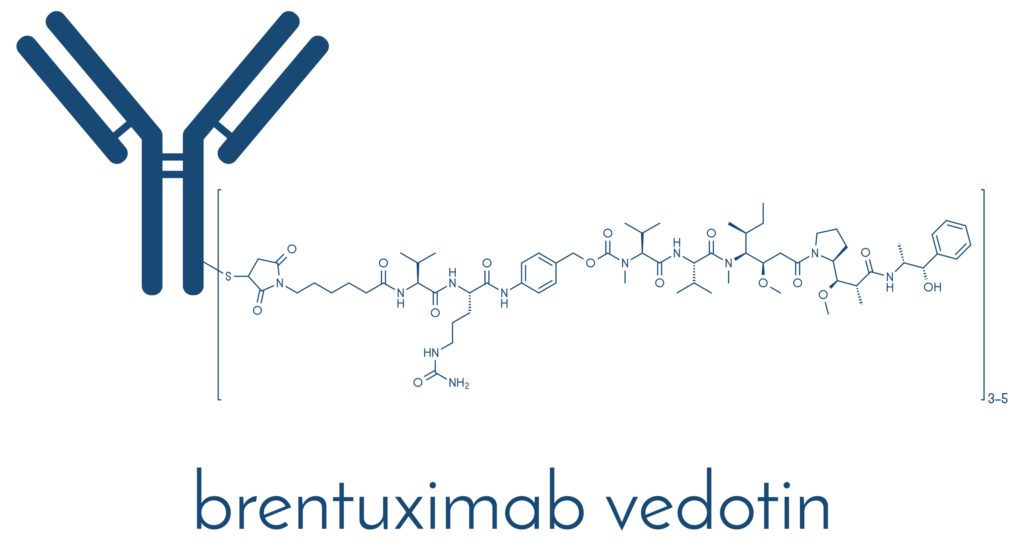
When we take brentuximab vedotin (1) as an example, it is an antibody-drug conjugate against CD30. Here, cAC10 is conjugated to SGD-1006 using thioether bonds. Therefore, the ADC has three key components, a chimeric IgG1 antibody cACIO that is specific for hCD30, an anti-microtubule agent monomethyl auristatin E, as well as a cleavable linker that attaches the two with covalent bonds. As one can imagine, the sequencing and engineering of the components can affect human biodistribution and pharmacokinetics of the ADC. That may very well dictate the safety and efficacy of the ADC. In this context, something drug developers pay attention to early on is the drug-to-antibody ratio. From a discovery standpoint, one hopes for a highly effective (i.e., potent) payload with a high drug to antibody ratio. Of course, another critical aspect is the linker, where the goal is to find a tumor selective linker to improve targeting as well as one that is stable in the systemic circulation.
Develop bioanalytical assays
One might think that developing bioanalytical assays is as simple as characterizing the pharmacokinetics of any new molecule. ADCs have both small molecule and large molecule components. Therefore, this leads to a very peculiar characterization of the clinical pharmacology profile. pay attention to the analytic species that need quantification.
Analytes may include the conjugated antibody, conjugated payload, total antibody (conjugated plus unconjugated), as well as unconjugated antibody and unconjugated payload (or the “free payload” in small molecule terms). Confusing? What is more confusing perhaps is that a single analytic method for all of them is quite unlikely. So, each method used will need to undergo qualification and validation throughout the development continuum.
So, what are we measuring? It is simple. We have an engineered 3-component new molecule. Thus,
- total antibody is the best reflection of antibody-related pharmacokinetics of the ADC
- the conjugated antibody reflects the active ADC concentration
- the conjugated drug is an estimate of the active drug that is associated with the antibody, and
- the free drug reflects the potency of the drug species and any impact on toxicity.
Don’t forget the small molecule!
Most makers of ADCs are biotechnology enterprises with strong expertise in biologics development who may not fully appreciate the nuances of the small molecule development. And of course, vice versa! This isn’t a reflection of their expertise, but rather a reflection of the inherent differences between biologics and small molecules nonclinical and clinical characterization. An extensive battery of preclinical and clinical testing is required for the small molecule component, including but not limited, to drug interaction assessment, specific populations, and considerations of other intrinsic and extrinsic factors that may affect the pharmacokinetics and biodistribution of the linker and the payload. The most proximal concern is that the cytotoxic payloads, when released from the ADC, behave like a typical small molecule and therefore are subject to a wide variety of influences by drug metabolizing enzymes and transporters, and perturbations thereof. So, what is clearance of an ADC? It is the combination of two parallel processes, loss of drug from the ADC and the catabolism of the ADC itself.
Therefore, understanding of the absorption, distribution, metabolism, and excretion (ADME) process of ADCs, elimination pathways, specific population studies, drug interactions and any covariates that might alter the pharmacokinetic exposures of these payloads are just a few key considerations. In the case of the antibody component, they undergo deconjugation to an unconjugated antibody, catabolized to amino acids and undergo elimination. The types of drug development questions that typically come up include, but is by no means an exhaustive list, stability of the linker, tissue distribution sites, potential for active and toxic metabolites, drug interaction potential, altered biodistribution, species specificity or lack thereof, QT prolongation potential of the payload (don’t forget the hERG cardiac safety test!), and of course, the bread and butter translational challenges of dose selection.
All said, the choice of the linker and the payload obviously depends on potency and safety considerations. Most employed cytotoxins are those that are naturally derived or are their synthetic equivalents, and ones that have a defined effect on the microtubules (e.g., maytansinoid or auristatin derivatives). The linker that connects the cytotoxic payload to the antibody is equally, if not more important, than the payload itself, as they need to be stable in the systemic circulation to release the payload at the desired site of action, which is the cytosol in the target cell. Premature release would mean contending with systemic safety considerations.
Translational strategies
This is perhaps the most important development consideration as it relates to the specificity and targeting necessary to achieve apoptotic cancer cell death, without systemic toxicity. Given the intent of the ADC, focus has been given to the expression of the target receptors on the tumor cells. There is an implicit consideration of the therapeutic window due to the highly potent payload and a risk of going “high” on dose because of that. Collection of biomarkers to characterize the mechanism of action, widening the therapeutic margins (ensuring dose is not unduly high), or assisting in choice of the target patient population, are just a few of the examples of potential impact.
The work of Fuh et al is a good example of leveraging biomarkers to characterize ADCs’ mechanism of action and pharmacokinetics (2), wherein the authors evaluated the ability of anti-CD22 and anti-CD79b ADCs to deplete proliferating cynomolgus monkey B cells. In that regard, they measured concentrations of peripheral blood Ki-67+ B cells as a biomarker of interest. Their findings revealed that the ADCs resulted in an extended duration of B-cell depletion in blood when compared with unconjugated antibodies. Interestingly, this effect was achieved despite the overall lower systemic exposure of ADCs, concluding that the difference in pharmacodynamic effects between the ADC and the unconjugated antibodies were mediated by a differential MOA, not necessarily the exposure. What makes this example pertinent to this discussion is that biomarker utilization not only showed value for mechanism of action, but also to explain the differences in PK/PD profile. Certara scientists have considerable experience in complex biologics including ADCs. If we can assist with your development needs, please contact us.
参照文献
- Brentuximab vedotin summary basis of approval (US FDA). URL: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125388_adcetris_toc.cfm (Accessed 2021年6月22日).
- Fuh FK, Looney C, Li D, et al. Anti-CD22 and anti-CD79b antibody-drug conjugates preferentially target proliferating B cells. Br J Pharmacol 2017;174:628–40.

