
Editor’s Note: Below are responses to the questions submitted by members of industry during GlobalSubmit’s Module 1 Education Series – M1 Effect on Ad Promo Submissions Webinar on 2015年6月16日.
1. Now that industry is able to submit OPDP submissions in eCTD format, can you foresee any issues/challenges when a sponsor and a partner marketing company are working concurrently? For example, the sponsor is creating and submitting all the usual lifecycle submissions in their tool and through their ESG account. The partner marketing company is compiling all the OPDP eCTD submissions using their own eCTD tool and submitting to the same application through their own ESG account?
If the sponsor and partner marketing company are working on the same application and submitting to OPDP in eCTD, then both companies will need to use the same version (us-regional-v3-3.dtd) of M1.
A potential complication to be cognizant of when compiling submissions as part of a collaborative agreement for promotion is duplicating sequence numbers. In the FDA’s guidance document, the Agency recommends devising a system where a block of numbers is set aside and assigned to each organization. For example, the partner marketing company would start promotional submissions with sequence 5000.
Duplicate sequence numbers would result in rejection of one of the submissions.
2. Is there a file size limit?
The file size is set at a maximum limit of 400 MB per the eCTD specification. Validation error 1238 deals with files that exceed the recommended limit. The 400 MB limit may not be applicable to datasets.
3. Are we limited from submitting grouped submissions across NDA/BLA for just Ad Promo submissions, or does that apply to all grouped submissions?
Limitations apply to all grouped submissions, including Ad Promo. Here are the main rules governing grouped submissions that you should be aware of:
- Application type (ex: NDA) has to agree for grouped submissions. For example, an NDA application type cannot be grouped to a BLA application type. A piece of promotional material that applies to both an NDA and BLA would have to be submitted separately.
- Submission type (ex: Labeling Supplement) has to agree for grouped submissions
- Grouping is not allowed across applications to CDER and CBER
- Grouped submissions are only supported using DTD version 3.3 or higher. When you choose to use the new M1 specifications at the start of a regulatory activity, you cannot regress to an older version.
4. Are grouped submissions the same as TRANS-BLA?
A trans-BLA was a grouped submission before grouped submissions were enshrined in the M1 specification.
For anyone unfamiliar with the term, a trans-BLA was a CBER concept. A single lead application with content would be chosen and that content would be applied to each BLA affected administratively. The process obviated the need for separate identical submissions, eliminating duplicate data entry, and enhancing efficiency.
The introduction of grouped submissions allows for multiple BLAs to be grouped formally using the new technology and administrative functionality. Content for each affected BLA will now be fully displayed.
5. We understood that sequence number was eliminated from your last presentation, however, today we saw a presentation showing sequence number. Please advise.
Sequence number and related sequence were not done away with – the FDA has merely modified how the information is collected.
Specifically, the sequence number concept has not changed. The number must be unique within the application and includes 4 digits.
What has changed is the FDA’s methodology for collecting and organizing metadata associated with each application. The old M1 specifications arranged information in a flat structure, i.e., amendments could be related to other amendments. In the new M1 specifications, metadata is arranged in a hierarchy.

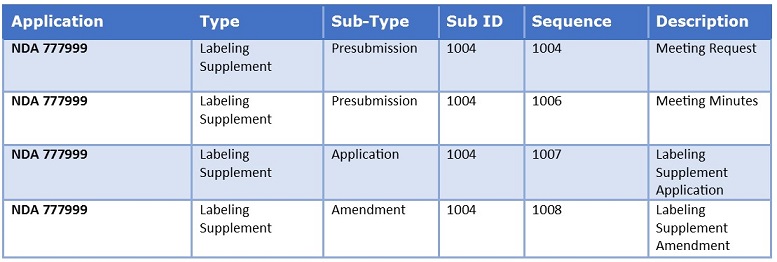
The first sequence number for each new “regulatory activity” is a new submission type, for example, a labeling supplement. That first sequence number for a new regulatory activity becomes the submission ID and the submission ID remains constant to relate submissions to a specific regulatory activity.
Take a look at this sample table to get a better idea of the relationship between Sub ID, sequence number, and regulatory activity.
6. Mandatory OPDP through eCTD? Two years from now? If I start using the new M1 now for grouped submissions I assume I could still do my OPDP on CD?
Correct. The new M1 specifications are only required if you are submitting to CDER OPDP in eCTD format. Promotional materials can still be submitted using paper and mixed media (CDs, DVD, etc.)
It is even permissible for a sponsor to use M1 specifications for grouped submissions (DTD v3.3) while continuing to submit promotional materials in paper for the same application.
7. Do we get receipt of confirmation from OPDP?
はい、対応しています。As part of the standard electronic submission process, sponsors receive a notification that each sequence has been received through the ESG, and a second notification after the submission has passed a technical validation check to ensure that it can be opened, processed, and archived.
8. When does the new M1 become mandatory?
The FDA has not announced a retirement date for us-regional DTD v2.01. The updates to the new M1 specification closely align to Regulated Product Submissions (RPS) or eCTD v4.0. New M1 specifications are more of a bridge to RPS than a permanent fixture. There is a good chance that the Agency will choose to jump straight to RPS implementation and never make DTD v3.3 mandatory. Of course that depends on the timeline for the RPS standard, which is currently in the process of being ratified as an international standard.
9. Can you please clarify the difference between an Amendment and resubmission?
The difference between an Amendment and Resubmission as taken directly from Providing Regulatory Submissions in Electronic and Non-Electronic Format – Promotional Labeling and Advertising Materials for Human Prescription Drugs, Section VI – Format for Submission of Promotional Materials Electronically, Subsection B – Submission-Type and Submission-Sub-Type:
Resubmission – Use this submission-type for requests for advisory comments and presubmissions of revised promotional materials that were previously submitted as an “original” submission.
Amendment – Use this submission-sub-type for a submission that contains additional supportive material to augment information previously submitted, e.g., the submission of promotional material that was previously missing or rejected, withdrawal requests, and submissions of annotated references. In addition, use this submission-sub-type for responses to untitled/warning letters, responses to information requests, and general correspondence if there was an original submission to FDA in eCTD format.
10. Do we need to file a sample submission with the new M1?
No. Sample submissions using the new M1 specification are not required. However, the FDA is accepting sample submission at this time.
11. In regards to an original 2253 submission, it appears that there was not a reference or link made to the current USPI. Is that correct?
When submitting 2253 submissions you should include the current product labeling under 1.14.6. Please refer to from Providing Regulatory Submissions in Electronic and Non-Electronic Format – Promotional Labeling and Advertising Materials for Human Prescription Drugs.
12. Will the FDA require the ID aka DUNS number?
At this time there is no validation check for the ID aka DUNS number and the FDA doesn’t have plans to use it, only collect it, at least as it pertains to eCTD submissions.
The DUNS number is marked as a mandatory field in GlobalSubmit’s eCTD publishing software application wizard.
13. Which is the main guidance document for Ad Promo submissions?
The guidance you should refer to for all matters Ad Promo submissions is titled Providing Regulatory Submissions in Electronic and Non-Electronic Format – Promotional Labeling and Advertising Materials for Human Prescription Drugs.
Click here for full text of the main guidance document
For reference, here are a few other presentations and documents you’ll want to familiarize yourself with, and hyperlinks directly to those resources.
Specifications for File Format Types Using eCTD Specifications
14. What if our partner company does not have the ability to use the new M1 but will do OPDP submissions?
If your partner company does not yet have the ability to submit using the new M1 specifications, perhaps their vendor’s technology isn’t compliant, OPDP submissions would have to be submitted to CDER in paper format. Again, the new M1 specifications are necessary when submitting promotional labeling and advertising materials to CDER OPDP in eCTD format.
15. Distinguish between Professional and Consumer audience if a website has content intended for both.
There are a few grey areas that exist for Promotional Material Audience Type, and we recommend familiarizing yourself with the main guidance document for Ad Promo – Providing Regulatory Submission in Electronic and Non-Electronic Format – Promotional Labeling and Advertising Materials for Human Prescription Drugs.
As far as a website with content for both professional and consumer, the classification largely depends on how the content is organized. Per the guidance:
“Websites with distinct sections for healthcare professionals and consumers should be divided into two separate submissions. If the website does not have distinct sections for each audience and is not intended to be directed solely to healthcare professionals, firms should submit the entire website as a consumer submission.”
