
治験薬 (Investigational New Drug 、IND) の初回申請または臨床試験開始申請(CTA)、新薬申請 (New Drug Application、NDA)、生物製剤承認申請 (Biologics License Application、BLA)、医薬品販売承認申請 (Marketing Authorization Application、MAA) 、その他の市場化申請作業は、大変な取り組みです。承認申請の成功には、多様な分野の専門家で構成された大規模なチーム、多数のツール、計画策定、各々のプロセス、その全てが互いに連動することが不可欠です。複雑性、リスク、時間、コストといった言葉は全ての承認申請の本質を表していると言えます。慎重に計画、実行し、予期せぬ問題を乗り切る力がなくては、薬事申請は失敗する可能性があります。
Keep your submission on track with Certara’s dedicated submission leads who have extensive experience advising clients and leading projects to submission and approval. Our submission leads all have more than 20 years of experience in significant submission leadership roles for biopharmaceutical companies. Supporting them is a pool of more than 200 expert project managers, regulatory writers, editors, and publishers, allowing us to build the right team for your product and submission.
“You may have seen the announcement that the FDA has accepted our NDA filing. I just want to say, thank you to each of you and everyone else at Certara who helped us get to this point. There’s more to come, but this is a big deal for us and soon for patients. We couldn’t have done it without you.” –Vice President, Program Management at a biopharmaceutical company
頑健な承認申請を実現する比類なき専門性
- More than 96% of 300+ submissions were approved
- 過去5年間に手がけた252件の承認申請の100%が予定よりも前に納品
- Supporting more than 50 submissions per year now across all submission types, therapeutic areas, product types, and geographies
- Expert in designing, planning, and delivering to increasingly expedited submission timelines with parallel authoring
- 迅速承認、緊急使用許可、ファストトラック指定、優先医薬品(PRIME)、および画期的治療薬指定といった制度の対応
- Extensive experience with rare/orphan disease submissions (64 in the past 5 years)
- 各国の規制要件を考慮したグローバル承認申請の準備と主導的役割
- Planning, strategy, and delivery expertise on simultaneous submission across US, EU, and Canada
- Experience in leading successful submissions in most major markets, as well as South America, Africa, and Eastern Europe
As key opinion leaders, Certara’s submission leads speak regularly at major industry events such as DIA Annual and AMWA’s Medical Writing and Communication Conference.

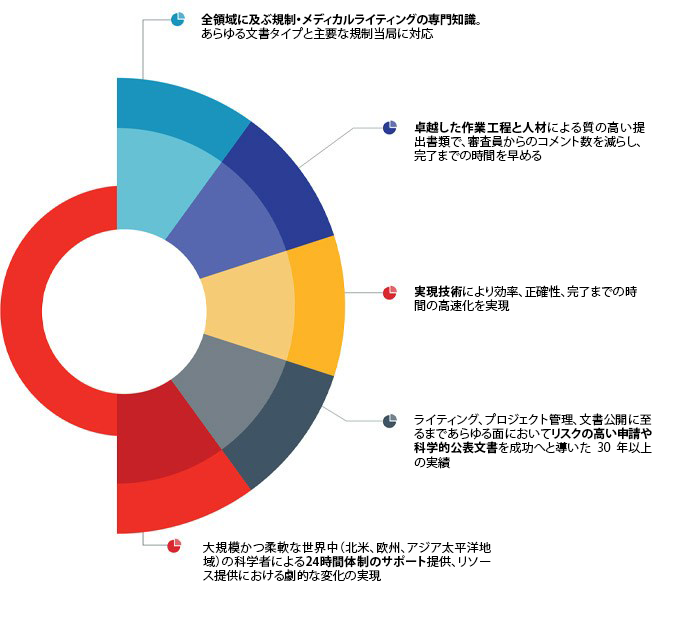
承認申請の迅速化に導くレギュラトリー
ライティング
従来のメディカルライティングや規制対応に特化したレギュラトリーライティングの分野において高い専門性を備えるサターラのライターチームには、あらゆる部門(文書品質管理、CMC、非臨床、臨床、薬事など) 出身の、博士、医学博士、薬学博士、 科学者、医療従事者などの経歴を有するエキスパートが所属しています。
Industry best-in-class SOPs drive quality, while dedicated QC functions and integrated writing teams reduce reviewer comments and speed time to completion.
承認申請成功に向けたサポート
Our submission leads and project managers are experts in applying project planning and management tools to accelerate and ensure successful delivery, with a state-of-the-art SmartSheet timeline and content plan tool for tracking and reporting progress.
- 添付文書やストーリーボード、スタイルガイドなど、全般的な文書作成方針を早期に策定することで、申請文書全体にわたって一貫性を保持
- 規制当局に自社開発品やデータを最善の方法で提示するサポート
- データセットや症例報告書 (Case Report Form:CRF) に関する内容や形式の議論も含めた、規制当局との事前相談のサポート。


医薬品業界での 30 年余にわたるキャリアを持つ、Steveは、レギュラトリーライティング、コンサルティング、プロジェクトリーダーシップの職務において、豊富な経験を有しています。彼は創薬から薬事承認まで、またライフサイクル管理において、多くのプロジェクトの成功を支えてきました。具体的には、75件以上の薬事申請において重要な役割に就き、いくつかの事例では薬事申請チーム全体を指揮し、モジュール1から5までのすべての書類作成、文書公開、規制当局への送信を監督してきました。

Dr. Bowlbyは臨床研究および医薬品開発業界において25年余の経験を有しています。直近の10年間は、米国食品医薬品局および欧州医薬品庁への数多くの新薬申請、生物製剤承認申請、治験薬(IND)申請をリードしてきました。また、治験薬概要書(IB)、 治験総括報告書(CSR)、臨床試験の要約および概要、対面説明資料(briefing package)、およびその他の規制文書のオーサリングも指揮してきました。それ以前の職務では、ハーバード・メディカル・スクールにおける自身の科学的研究と博士課程修了後の研究に関して、多数の生物医学的な原稿、ポスター、スライドを計画・執筆しました。彼の専門の治療領域には、眼科、神経内科、精神科、慢性疼痛管理分野が含まれます。最新の生物医薬品環境で使用されている創薬および医薬品開発アプローチについての専門的知識を有しています。
医薬品業界での 30 年余にわたるキャリアを持つ、Steveは、レギュラトリーライティング、コンサルティング、プロジェクトリーダーシップの職務において、豊富な経験を有しています。彼は創薬から薬事承認まで、またライフサイクル管理において、多くのプロジェクトの成功を支えてきました。具体的には、75件以上の薬事申請において重要な役割に就き、いくつかの事例では薬事申請チーム全体を指揮し、モジュール1から5までのすべての書類作成、文書公開、規制当局への送信を監督してきました。
Dr. Bowlbyは臨床研究および医薬品開発業界において25年余の経験を有しています。直近の10年間は、米国食品医薬品局および欧州医薬品庁への数多くの新薬申請、生物製剤承認申請、治験薬(IND)申請をリードしてきました。また、治験薬概要書(IB)、 治験総括報告書(CSR)、臨床試験の要約および概要、対面説明資料(briefing package)、およびその他の規制文書のオーサリングも指揮してきました。それ以前の職務では、ハーバード・メディカル・スクールにおける自身の科学的研究と博士課程修了後の研究に関して、多数の生物医学的な原稿、ポスター、スライドを計画・執筆しました。彼の専門の治療領域には、眼科、神経内科、精神科、慢性疼痛管理分野が含まれます。最新の生物医薬品環境で使用されている創薬および医薬品開発アプローチについての専門的知識を有しています。
